Indice dei contenuti
Descrizione generale della malattia
È una malattia genetica
I tre geni principali che causano la CMA fra quelli finora identificati codificano per tre diverse proteine, (Plakofilina, Desmoplakina e Plakoglobina), che formano i “contatti” fra una cellula e l’altra del cuore, denominati giunzioni intercellulari o anche “Desmosomi”.
In conseguenza delle mutazioni genetiche queste proteine sono difettose e possono provocare le rotture delle giunzioni fra cellula e cellula e quindi la loro scomparsa, sostituite da tessuto fibrotico cicatriziale associato a dilatazione localizzata o diffusa ventricolare.
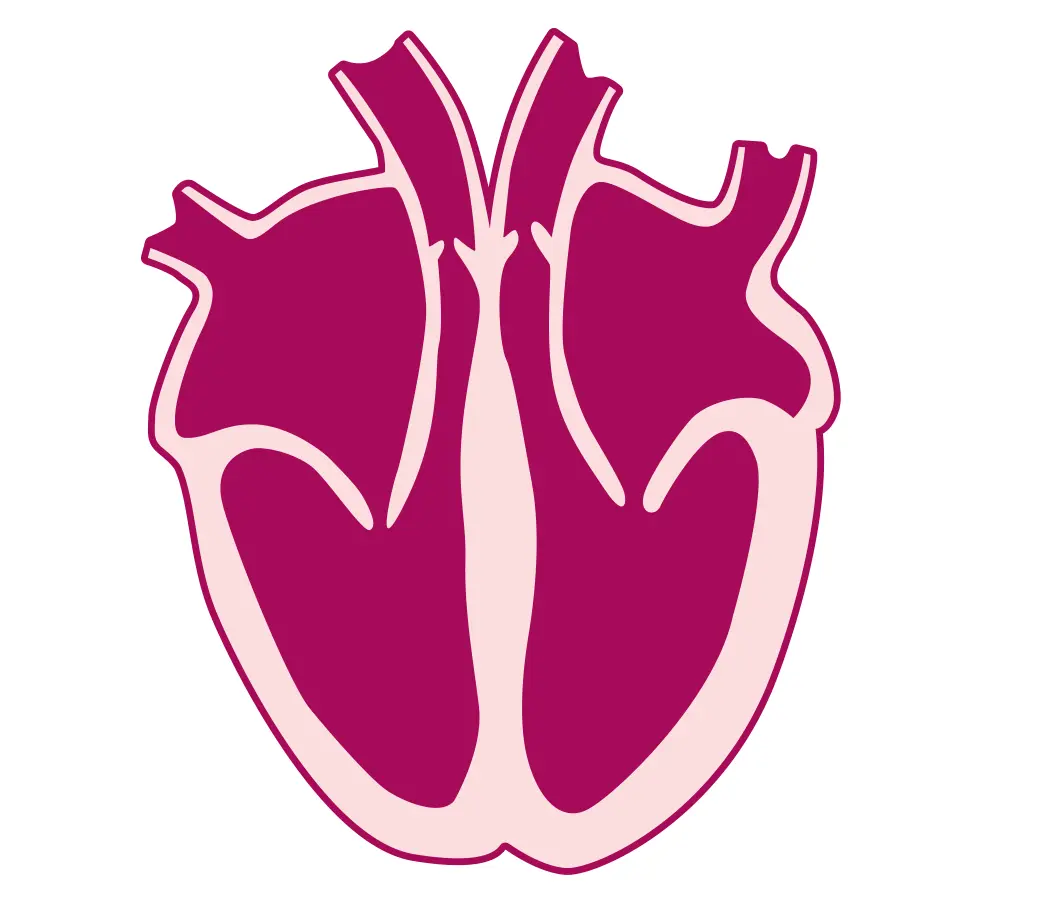
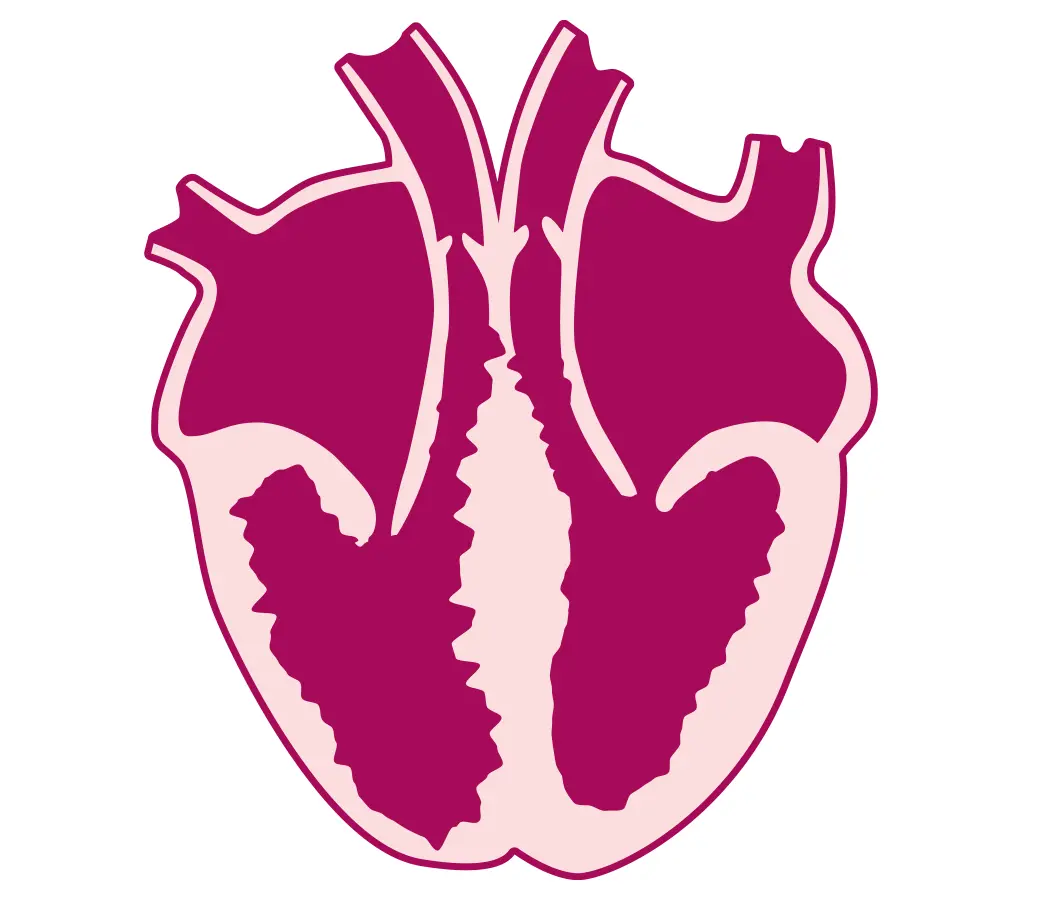
Cardiomiopatia aritmogena destra e sinistra: fibrosi delle pareti, aumento di cavità e ridotta forza contrattile del Ventricolo destro e/o sinistro, con associate aritmie ventricolari anche minacciose (Tachicardia ventricolare o Fibrillazione ventricolare)
Sintomi
Inizialmente la CMI è spesso asintomatica, ovvero i sintomi possono essere assenti o non percepiti dal paziente e la diagnosi in questi casi viene fatta per controlli casuali come ad esempio in occasione di una visita medico sportiva. I sintomi più comuni, quando presenti, sono:
Diagnosi
Riconoscere e fare diagnosi di CMA non è facile, in particolare quando le anomalie sono in fase iniziale e limitate ad alcune zone, difficilmente. Un’attenta valutazione dell’ECG e delle pareti del ventricolo destro con Ecocardiografia deve far sospettare la diagnosi. Il sospetto spesso nasce poi in presenza di aritmie, battiti prematuri (extrasistoli) atriali e ventricolari, spesso scatenate da sforzo.
In alcuni pazienti aritmie più complesse (Tachicardia ventricolare sostenuta o Fibrillazione ventricolare) possono essere minacciose per la vita, determinare perdite di coscienza improvvise (sincopi) e/o portare all’arresto cardiaco.
Diagnosi
Per formulare la diagnosi è necessario eseguire esami clinici e strumentali non invasivi:
Successivamente vengono effettuate altre indagini, che includono:
Dalla Videoteca del Cuore
Approfondimenti video sulla cardiomiopatia aritmogena
Vedi anche le seguenti sezioni

Hai bisogno di aiuto?
Se tu o un tuo familiare avete ricevuto una diagnosi di Cardiomiopatia Ipertrofica, AICARM è al tuo fianco.
Il nostro servizio “Cuori in Ascolto” offre informazioni, orientamento e supporto a chi convive con questa patologia.