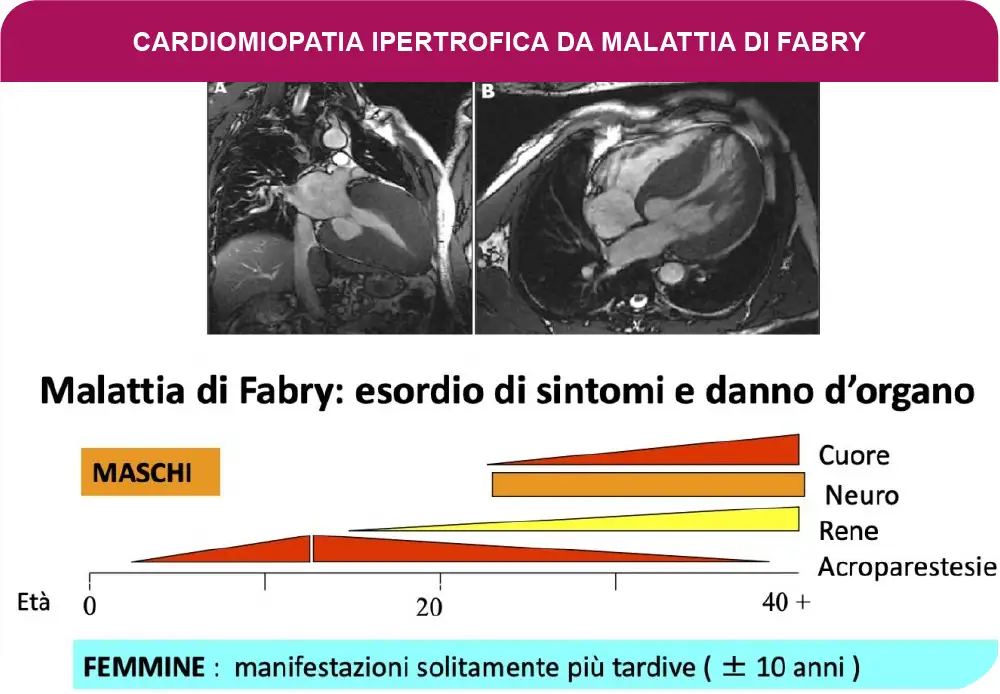
Se l’enzima è assente la malattia di Fabry si manifesta nei maschi già nell’infanzia e può determinare danno renale, cerebrale o cardiaco, già in giovane età. Se l’enzima è solo marcatamente ridotto, le manifestazioni cliniche compaiono più tardivamente, di solito in misura nettamente minore ed ancora più tardiva nelle femmine. Dopo i 20 anni nei maschi, ma soprattutto in età adulta ed anche avanzata, in particolare nelle femmine, i pazienti possono presentare una Cardiomiopatia Ipertrofica (CMI), con ipertrofia progressiva, anche di grado severo, isolata o associata a gradi variabili di insufficienza renale o di altri organi.
E’ documentato che circa 1-5% di pazienti di età superiore a 40 anni la CMI è dovuta a m.di Fabry.
Diagnosi:
viene effettuata misurando su sangue fresco l’attività dell’α-galattosidasi A leucocitaria con successiva conferma, se l’attività è ridotta o assente, mediante analisi genetica delle mutazioni sul gene specifico (GLA). Nei maschi la diagnosi di malattia di Fabry viene esclusa se i valori di attività enzimatica sono normali o solo lievemente ridotti. Tuttavia le femmine affette possono avere valori di attività enzimatica normali, ed è preferibile effettuare subito l’analisi genetica, il cui risultato va confermato come causa della malattia di Fabry da genetisti esperti.
Infatti il solo riscontro di una mutazione genetica non è sufficiente per fare diagnosi di m. di Fabry.
L’attività enzimatica dell’α-galattosidasi A leucocitaria e l’analisi genetica vengono effettuate, su appuntamento, in pochi laboratori specializzati in Italia (es. Ospedale Meyer a Firenze).
Tuttavia è anche possibile utilizzare uno specifico cartoncino sul quale si fa essiccare una goccia di sangue, chiamato DBS (dried blood spot), che va inviato nei laboratori che effettuano l’analisi in Italia o in Europa e può essere conservato fino a 3 mesi.
Terapia:
da circa 20 anni è possibile somministrare per via endovenosa l’enzima assente, efficace soprattutto nelle fasi non avanzate della malattia. Da alcuni anni è disponibile anche una terapia orale, da utilizzare quando l’attività enzimatica è solo ridotta. Sono allo studio anche forme di terapia genica.
In Italia esistono numerosi centri di riferimento specializzati nella diagnosi e terapia della malattia di Fabry.